Highly accurate protein structure prediction with AlphaFold
Highlights
- AlphaFold2 is an end-to-end deep learning network for predicting the 3D structure of proteins from the sequence of amino acids.
- The model won the 14th Critical Assessment of protein Structure Prediction (CASP14) challenge in 2020 by a large margin, acheving performances never seen before.
- The work of this team was recently awarded the Nobel Prize in chemistry (awarded to John Jumper and Demis Hassabis from Google DeepMind, as well as David Becker) for having “cracked the code for proteins’ amazing structure”.
- The paper, published in Nature, is rather short by itself, but all the details about the model, the training procedure and more are available in the 62-page-long supplementary material.
- The code is openly available on a GitHub repository.
- This video is of great use to understand in detail AlphaFold2.
Introduction
Understanding the 3D structure of proteins, these key building blocks of life on Earth, is essential for biologists. The sequence of amino acids constituing a protein is not sufficient to know its function, which is also greatly defined by the spatial arrangement of the molecule. Obtaining the 3D structure of a protein is feasible experimentally but is also incredibly costly and long : until now, the structures of around 100 000 unique proteins have been determined experimentally. However, up until a few years ago, predicting the 3D structure of a protein from its 1D sequence of amino acids remained an unsolved task. Such a task is of major interest as it has the potential to greatly accelerate research in biology and chemistry.
In 2020, Google DeepMind’s team called AlphaFold2 won the bi-annual CASP14 challlenge by a large margin over the other methods and achieved unprecedented accuracy, on par with experimental error. Two years before, DeepMind had already won the challenge, but the performance of the first version of their model was not as groundbreaking as its second iteration.

Figure 1. Example of the 3D structure of a protein obtained experimentally (green) and predicted by AlphaFold (blue).
Brief introduction to protein structure. A protein is a molecule composed of one or more polypeptides, that is linear chains of amino acid residues. Shorter proteins only contain few dozens of residues, but longer ones can have thousands of amino acids. An amino acid is composed of a “main structure”, or backbone of two carbon atoms, an amin group, a carboxyl group and a side chain, often called R (Figure 2). The different amino acids of a protein are bonded by a link between the amino and the carboxyl groups.
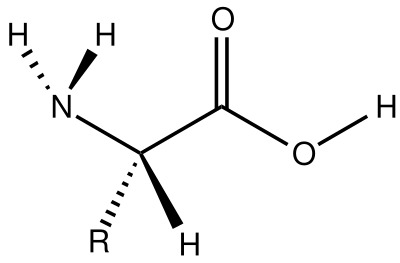
Figure 2. General structure of an alpha-amino acids composing proteins.
Motivation
The task solved by AlphaFold is quite far from our domain of reseach, image processing, but I do believe that reviewing this paper can be of interest for us, and not only for general scientific culture purpose.
AlphaFold2 is not only a feat of engineering, it also contains significant methodological contributions, and can be an inspiration for us in how we build deep learning based models for medical images, in particular showing the interest of adding appropriate inductive biases into the models.
Overview of AlphaFold
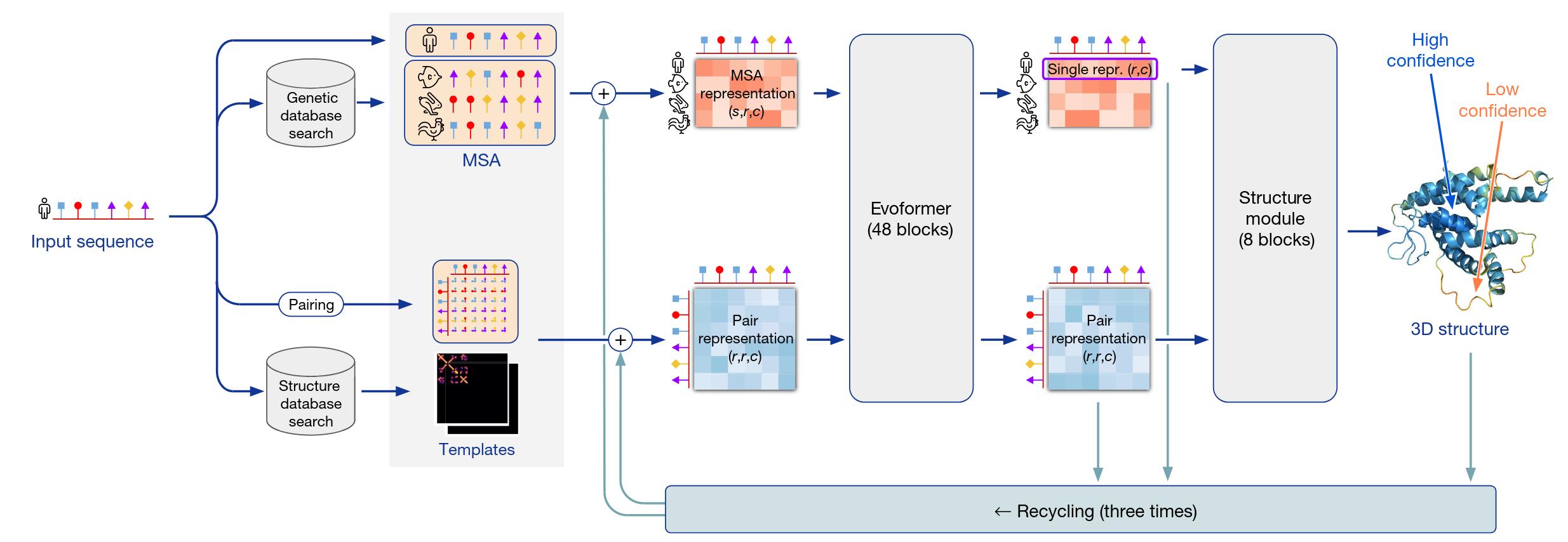
Figure 3. High-level overview of AlphaFold.
Let’s first see how AlphaFold works on a high level (Figure 3). It takes as input a 1D sequence of amino acids, more specifically of the 22 \(\alpha\)-amino acids that are present in proteins, and it aims to output the full 3D structure of the molecule, that is, the position and orientation in space of all its atoms. It combines two complementary approaches that have a long history in protein structure prediction that focus respectively on the physical interactions or the evolutionary history of a protein. Understanding the physical interactions heavily between the atoms can greatly guide the prediction of the structure of proteins via thermodynamic or/and kinetic simulations. However, these methods are usually computationally intractable and require accurately model the physic of the interaction inside the protein. On the other hand, evoolutionary approaches leverage the history of proteins, homologies between proteins of different species and pairwise cevolutionary correlations. It is allowed by the existance of the millions of protein sequences already available in the Protein Data Bank.
Deep dive into the architecture of AlphaFold
We will now present more in details some of the main components of AlphaFold. Due to the overall huge complexity of the model, we can not cover all its aspects and subtilities, but we hope that the following sections will help you understanding what makes the strength of the model.
Input embedding
From an input amino acid sequence, AlphaFold derives two main objets that will be used to predict the 3D structure : the Multi Sequence Alignment (MSA) matrix between the protein of interest and proteins with similar functions in different species and the pairwise representation between elements of the input sequence, embedded with features from template proteins (Figure 4).
The idea behind using the MSA matrix is to analyze the mutations that has occured inside amino acid sequence of proteins that share a common ancestor ad the same function. For instance, intuitively, when comparing the proteins of two species, if two amino acids always mutate at the same time, this can mean that there are really closeby in the 3D structure of the protein. Understanding the correlations in the MSA matrix is thus key to predict the spatial arragement of the target molecule.
The second type of objects processed by the model is the pairwise representation tensor between the elements of an protein sequence. Basically, you can think about it as an embedded version of the pairwise distances between all the atoms of the molecules. At this stage, the model also leverages template protein structure, which are the (embedded) pairwise distances of molecule for which we already know the exact 3D structure; it allows the model to look for similar structures.
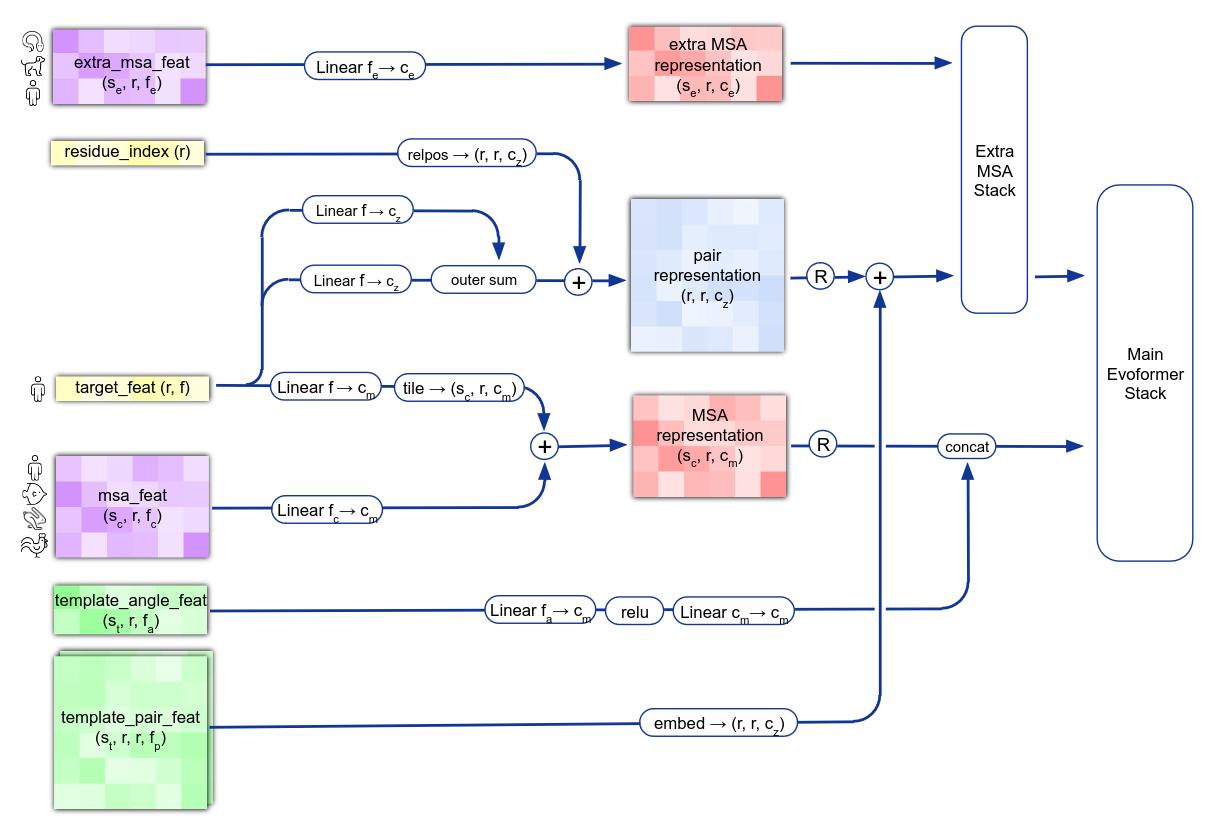
Figure 4. AlphaFold input embedding into MSA features and pairwise representation.
Evoformer
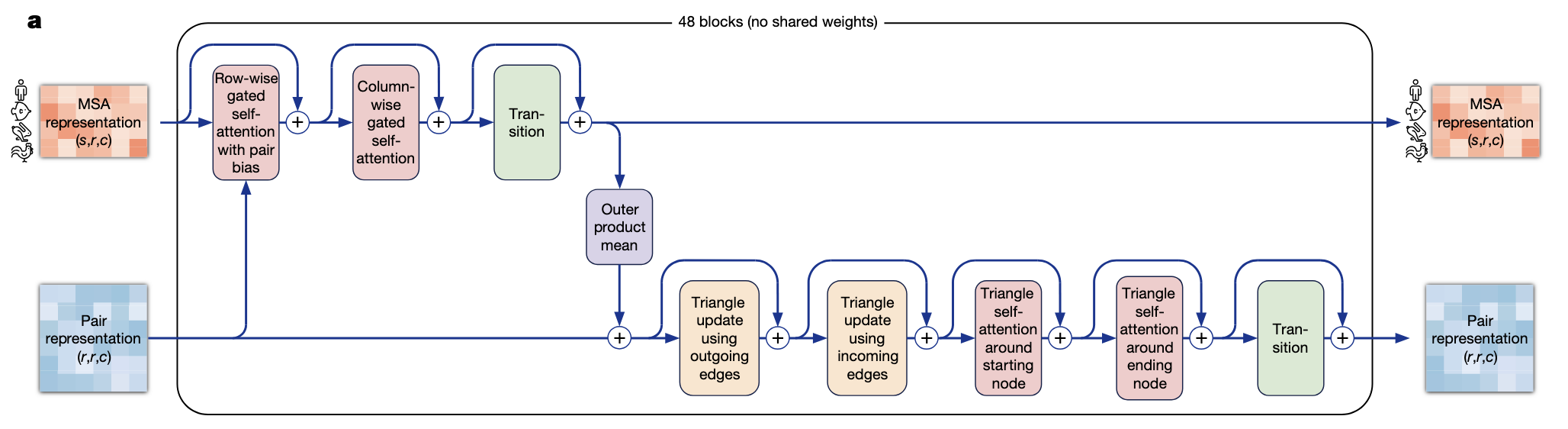
Figure 5. Structure of AlphaFold's Evoformer.
The MSA and pair representations are the two inputs of the first main component of AlphaFold, called Evoformer (Figure 5). It is a transformer-based model whose purpose is to update the MSA and pair representations with self and cross attentions.
One of the key ideas of this transformer is to incorporate clever inductive biases inside the operations performed by the model. As the number of dimensions of the inputs tensors is large – they can be seen as embedded 2D images –, instead of performing cross-attention on the flattened sequences of tokens, the MSA is modified via row-wise and column-wise self attention operations. Moreover, the MSA only contains feature information, without any information about the structure of the protein. To incorporate such knowledge into the MSA representation, the authors use a modified version of the row-wise self-attention where they add inside the softmax operation biases coming from the pair representation tensor (Figure 6). Note that this is only done for the row-wise operations and not the the column-wise ones as it does not really make sense to add this kind of structural information, specific to each protein, to a cross-attention of the information of one amino-acid between severel species (column-wise self-attention is basically the same as Figure 6 without the lower part of the schema).
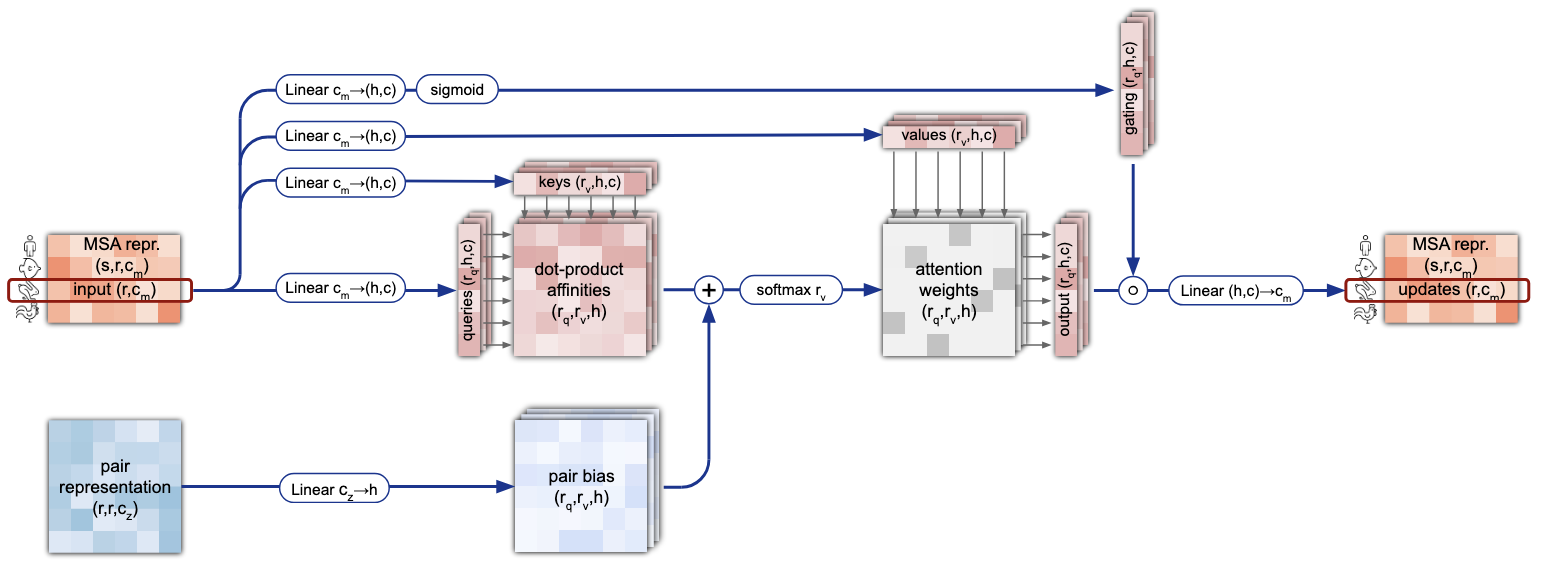
Figure 6. Details of the row-wise gated self-attention with pair bias.
The Evoformer also updates the pair representation, using element-wise addition with the updated MSA tensor followed by self-attention blocks. The paper also introduces very import novelty in these attention blocks. Their goal is to update the embedding of the structural representation of the molecule. Generic self-attention does not impose specific bias on the representation. However, if we think of the amino acid chain as a concatenation of small triangles (formed by the carbons and nitrogen of the backbone chain), one property that every triplet of points should satisfy is the triangular inequality : when considering a triangle, the length of a side cannot be greater than the sum of the lengths of the two other sides of the triangle. Such property, for the authors, is key when one tries to predict the shape of the 3D molecule. We can not directly impose such a condition in the network (plus we are dealing here with embedded positions, not strictly the Euclidean position of the atom for now), but the pair representation are updated with a modified self-attention whose goal is to mimic the spirit of the triangular inequality. For an edge \(e_{ij}\), we will consider all possible triangles with edges \(e_{ij}\), \(e_{ik}\) and \(e_{ik}\) (for \(k \neq i,j\)) and the edge \(e_{ij}\) will receive information from a combination of the two other edges of each of these triangles (Figure 7). The pair representation learns four similar self-attentions, respectively using “outgoing edges”, “incoming edges”, “starting node” or “ending node”.
Figure 7. Details of triangular multiplicative update of the "outgoing" edges.
In total, the Evoformer is made of a stack of 48 of the blocks depicted in Figure 5, without any weights shared between those blocks.
Structure module
At the end of the Evoformer, we get updated versions of the MSA and pair representations of the input sequence. The second main block of AlphaFold is called the Structure module. It aims to (almost) predict the final 3D structure of the input protein sequence.
The choice of how to represent the 3D final structure is a key architecture choice of the model. The author choose to predict the final position of the atoms in two steps : first, the predict the position of the main backbone of the protein chain, then the position of the side chain – remember, the side chain subpart we denoted R in each amino acid. Morevover, the composed te side chain as follows : they virtually “cut” the bond between each amino acids and model the (rigid body) amino acid by small triangular backbone frames, each triangle representing the position of the two carbons and the nitrogen of the amino acid (Figure 8).
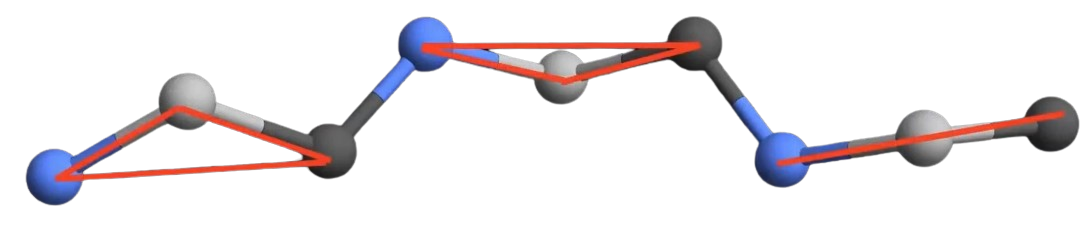
Figure 8. Backbone structure.
The position of all those triangles is initialized to the origin of the 3D space. For each triangle, the model will predict a 3D transformation \(T = (R, \vec{t})\) which is the composition of a translation \(\vec{t} \in \mathbb{R}^3\) and a rotation \(R \in \mathbb{R}^{3 \times 3}\) (in the referential of the atom).
The Structure module (Figure 9) takes as input the first row of the MSA, which corresponds to the representation of the target protein, the full pair representation and the transformations of the backbone frames (initially at the origin). It is composed of 8 blocks that shared the same weights : you can think about the prediction of the transformations of the backbone frames as an iterative process, where at the position is sequentially refined according to the previous position. The first iterations already achieve a satisfying general structure for the protein and the following refine it. Moreover, the structure prediction if refined by using the output of the network as an additional embedding for the inputs of the structure module. The procedure, called recycling in the paper, is done up to three times.
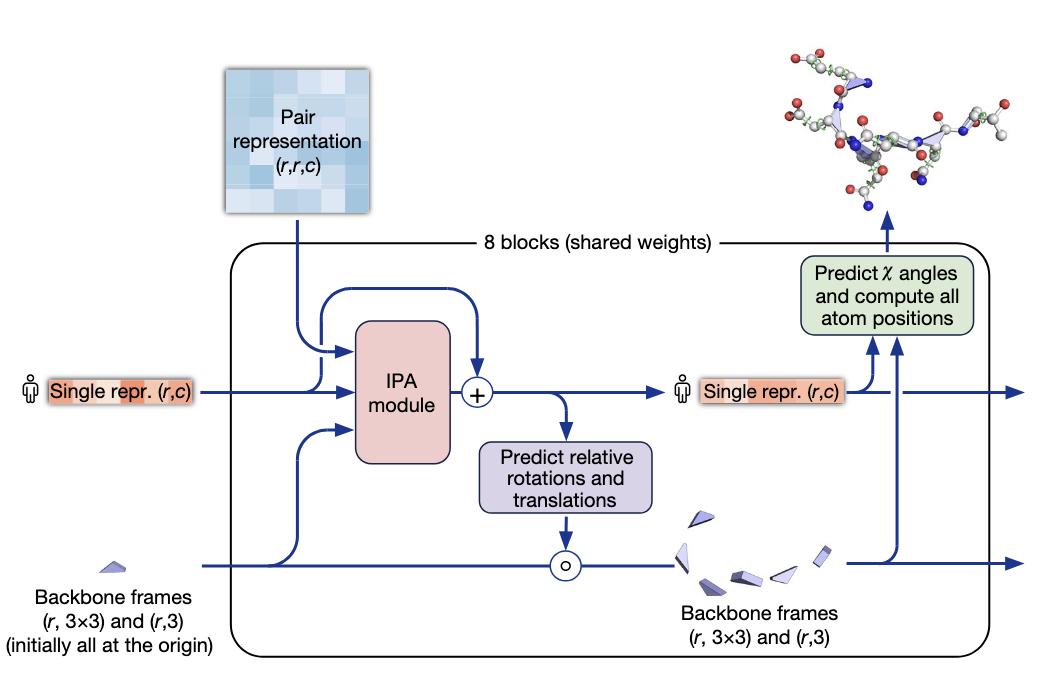
Figure 9. Details of the structure module of AlphaFold.
The main component of the structure blocks is the IPA module. Without going into much details, it updates the protein representation by embedding it with the pair representation and the global and local backbone frames coordinates.
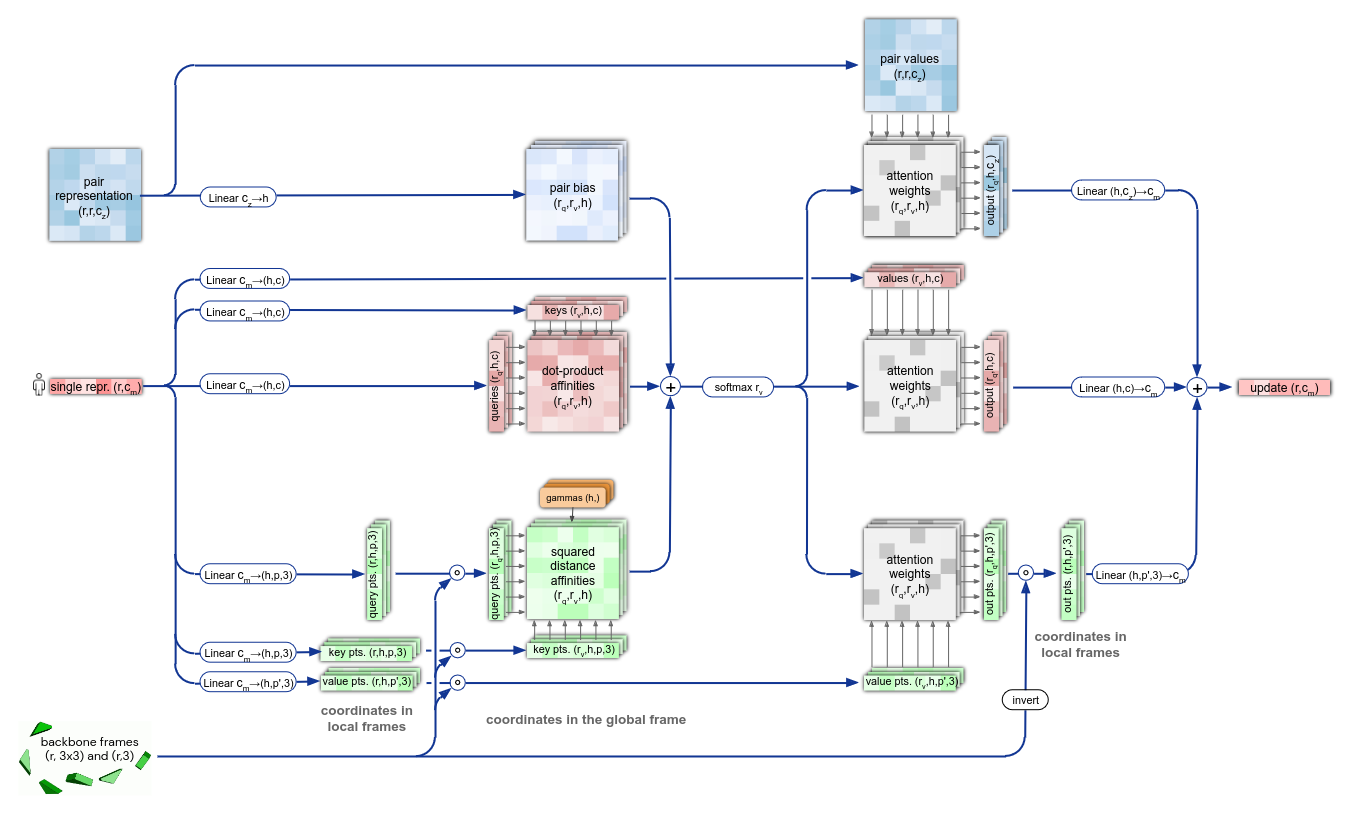
Figure 10. Details of the IPA module.
Training
The network is trained in an end-to-end fashion with a combination of multiple losses, shown below.
\[\mathcal{L} = \begin{cases} 0.5 \mathcal{L}_{\text{FAPE}} + 0.5 \mathcal{L}_{\text{aux}} + 0.3 \mathcal{L}_{\text{dist}} + 2.0 \mathcal{L}_{\text{msa}} + 0.01 \mathcal{L}_{\text{conf}} & \text{training} \\ 0.5 \mathcal{L}_{\text{FAPE}} + 0.5 \mathcal{L}_{\text{aux}} + 0.3 \mathcal{L}_{\text{dist}} + 2.0 \mathcal{L}_{\text{msa}} + 0.01 \mathcal{L}_{\text{conf}} + 0.01 \mathcal{L}_{\text{exp resolved}} + 1.0 \mathcal{L}_{\text{viol}} & \text{fine-tuning} \end{cases}\]We describe here some of the components of the loss function. The \(\mathcal{L}_{\text{FAPE}}\) loss (for Frame Aligned Point Error) is proposed by the authors and represents the error in the prediction of the global translation and local rotation of the triangular frames. One of its advantages is to solve the problem of chirality not taken into account with other losses, for instance when they are based on pairwise distances between atoms. \(\mathcal{L}_{\text{aux}}\) is a loss on the intermediate layers of the structure module (average of FAPE loss and torsion loss, not detailled here). \(\mathcal{L}_{\text{msa}}\) is a BERT-style loss computed on the MSA matrix at the output of the Evoformer : during the training, some parts of the MSA are masked and the network has to recover the missing parts of the MSA.
The training is done in two steps, an initial training and a fine tuning with noisy self distillation training. Simply put, after the first training, the resulting model is used on protein sequences for which the 3D structure is unknown and, if the confidence level for the predicted structure is high enough, it is included in a self-distillation dataset. The finetuning chooses samples from both Protein Bank Data and the self-distillation dataset, with a respective weight of 0.25 and 0.75. During training, the MSA is cropped to a size of 256 (384 for the fine tuning), and the number of sequences is fixed.
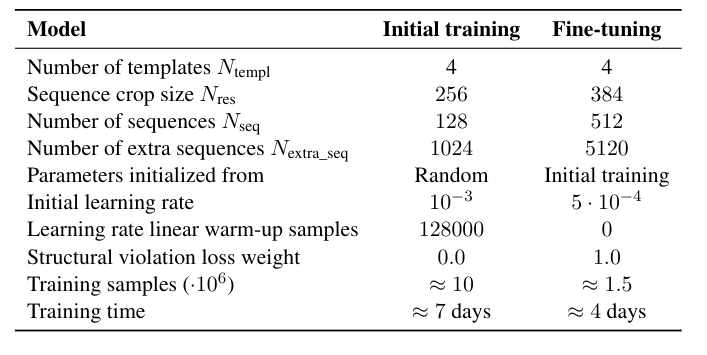
Figure 11. Training procedure of AlphaFold.
Main ablation study
The authors provide an ablation study of the main components and design choices of AlphaFold
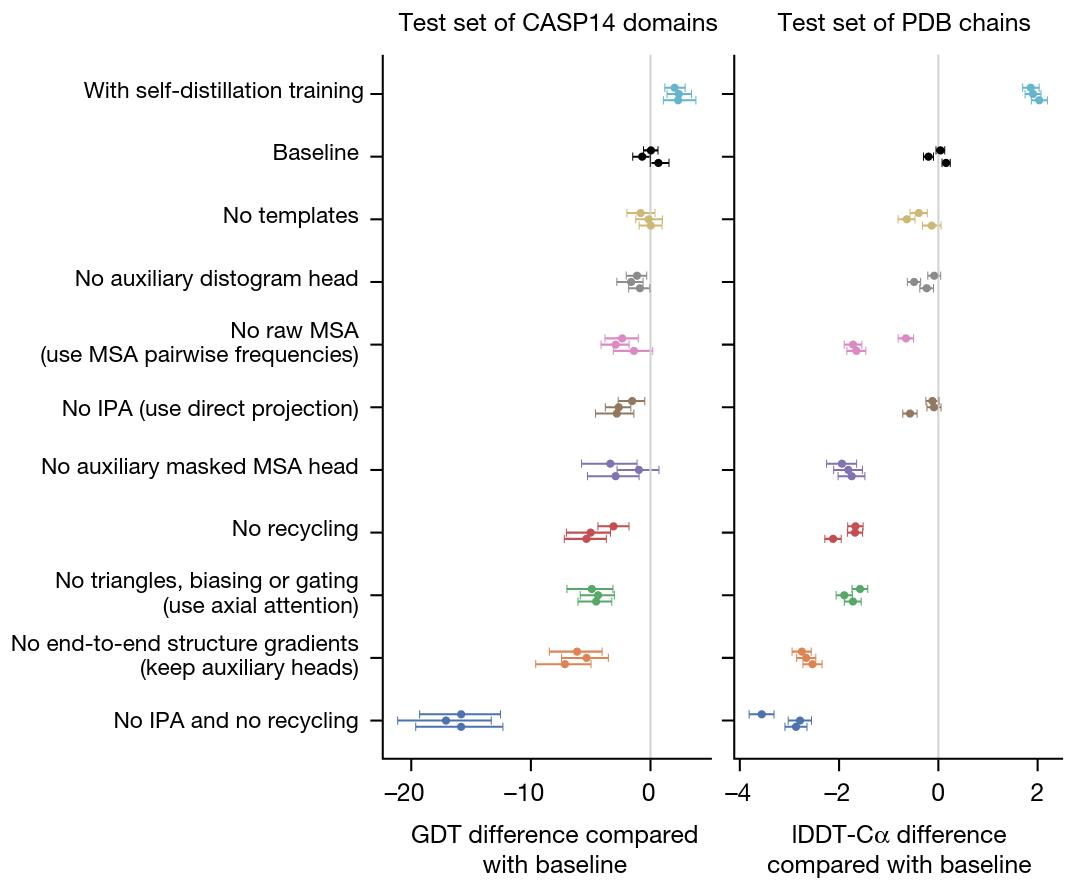
Figure 12. Ablation study of some of the components of AlphaFold.
Main results
The most important evaluation of AlphaFold’s abilities was done during the CASP14 challenge. The competing methods were evaluated on never-seen-before proteins and associated 3D structure, to ensure their generalization capabilities. The metrics used for the evaluation, such as the GDT score and IDDT-C\(\alpha\), are not discussed here, but the key takeaway message is the AlphaFold achieves a sub atomic precision, with a median r.m.s.d of 0.95 \(A\) (Angstrom) for the backbone atoms and of 1.5 \(A\) for all the atoms.
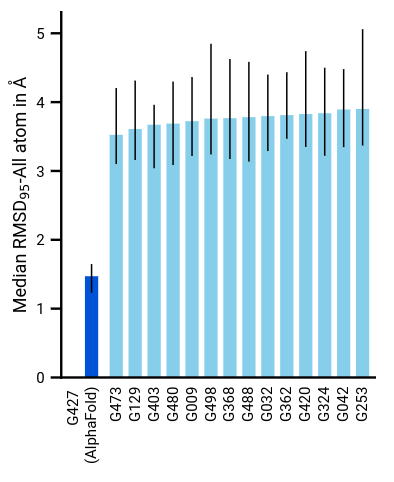
Figure 13. Median all-atom RSMD on the CASP14 set of protein domains (N = 87 protein domains) relative to the top-15 entries.
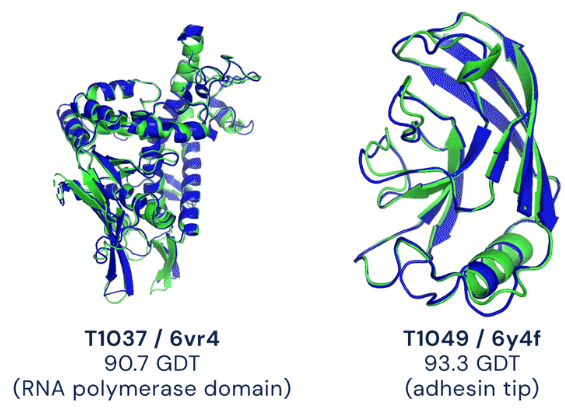
We showcase below some examples of predictions of 3D structure of molecules by AlphaFold, along with the ground truth obtained experimentally (in grey, or blue for the first two images). These images are actually not only proteins but also RNAs : they are predictions from the next iteration of Google Deepmind’s model, AlphaFold3, which can predict the spatial structure of a broader variety of biomolecules.
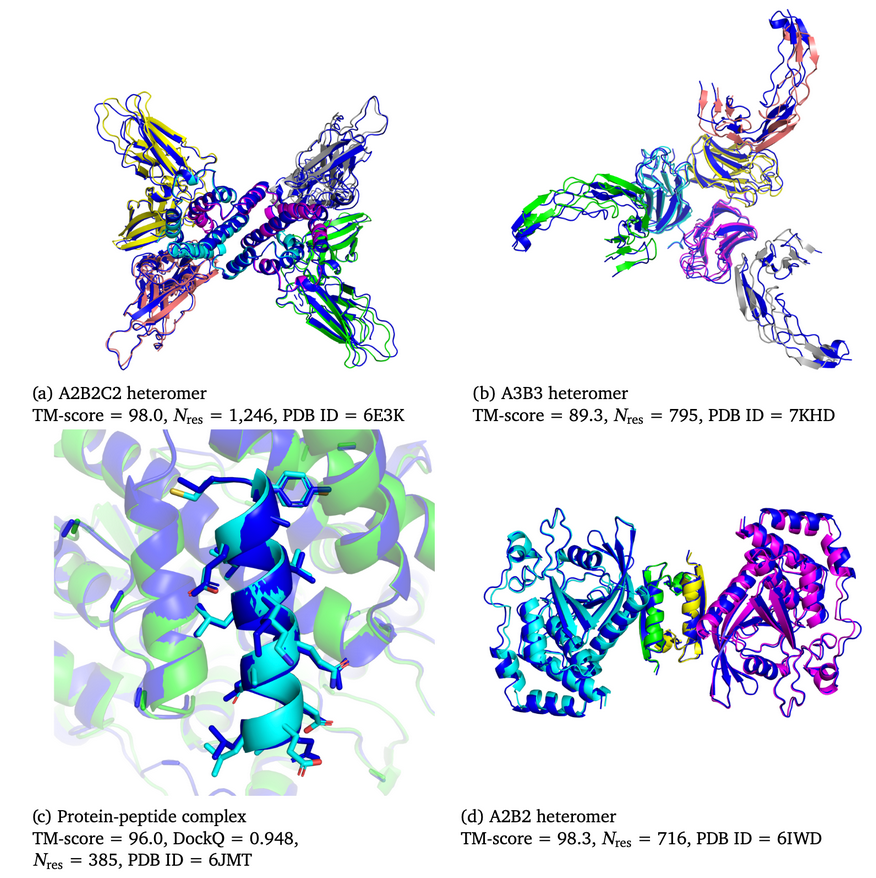
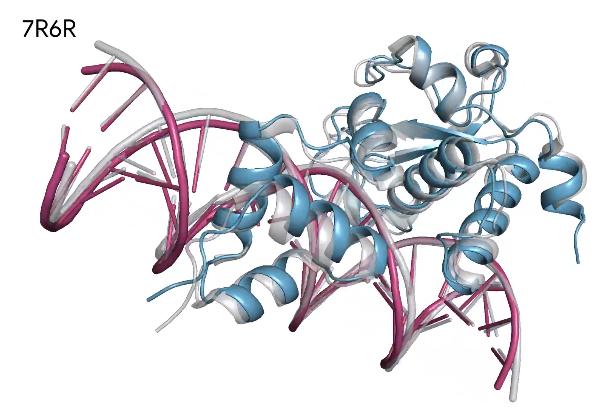
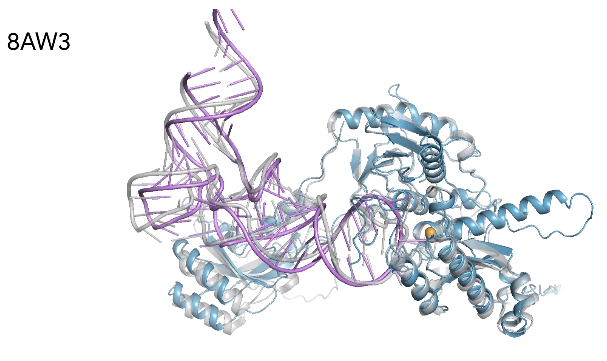
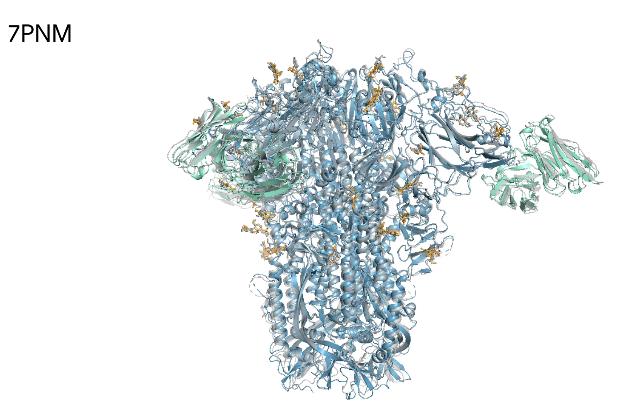
What’s next ?
Google Deepmind did not take part in any of the challenges that followed CASP14, but since then the best models have all been modified versions of AlphaFold2. As mentioned before, Deepmind has recently released, in collaboration with Isomorphic Labs, AlphaFold3, which can provide the spatial structure of a wider variety of biomolecules and with an improved accuracy compared to AlphaFold2. They have created a database with the structures of 200 millions of biomolecules predicted by their model, and allow any researcher to upload their own sequence to get a prediction from AlphaFold3.